

Google语音识别技术详解与实践应用

小杜的生信笔记,自2021年11月开始做的知识分享,主要内容是 R语言绘图教程 、 转录组上游分析 、 转录组下游分析 等内容。凡事在社群同学,可免费获得自2021年11月份至今全部教程,教程配备事例数据和相关代码,我们会持续更新中。
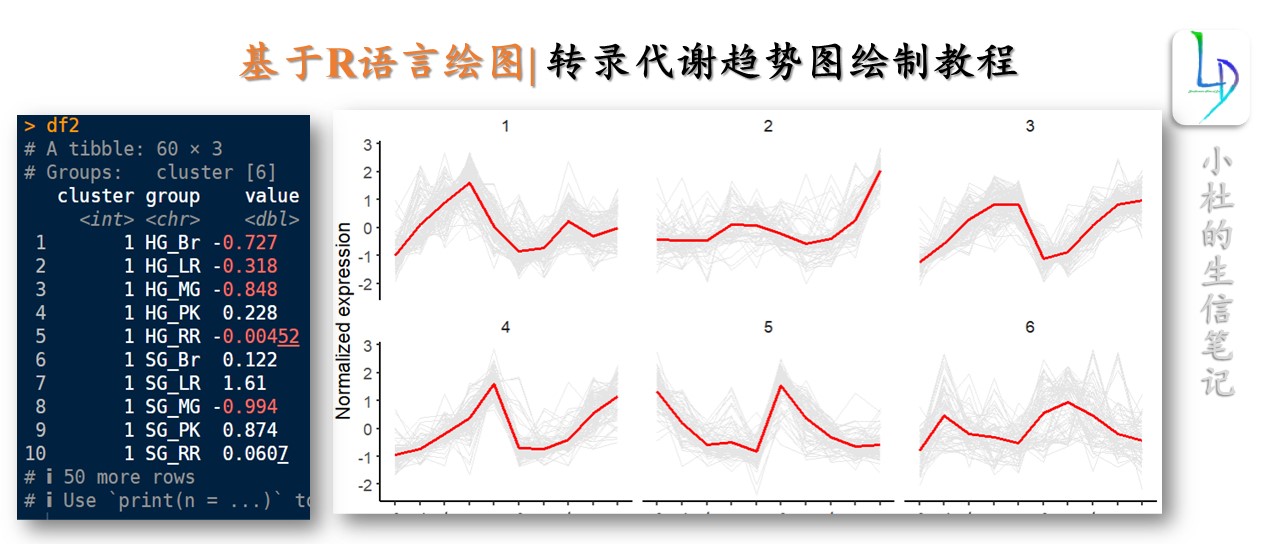
聚类分析用于将表达模式相同或相近的基因聚集成类,进而识别未知基因的功能或已知基因的未知功能,这些同类基因可能具有相似的功能,共同参与同一代谢过程或存在于同一细胞通路中。K-means称为K-均值聚类;k-means聚类的基本思想是根据预先设定的分类数目,在样本空间随机选择相应数目的点做为起始聚类中心点;然后将空间中到每个起始中心点距离最近的点作为一个集合,完成第一次聚类;获得第一次聚类集合所有点的平均值做为新的中心点,进行第二次聚类;直到得到的聚类中心点不再变化或达到尝试的上限,则完成了聚类过程。
在多组学分析中,趋势图是常见的图形之一,而聚类分析则是构建这类图形的基础。通过聚类,我们可以识别相似表达模式的基因,进而进行更深入的生物学解读。
ClusterGVisClusterGVis 包可以使用 k-means 或 mfuzz 进行聚类分析。具体操作,自己动手做一下即可。
一般,我们输入的都是 宽数据矩阵,如下所示:
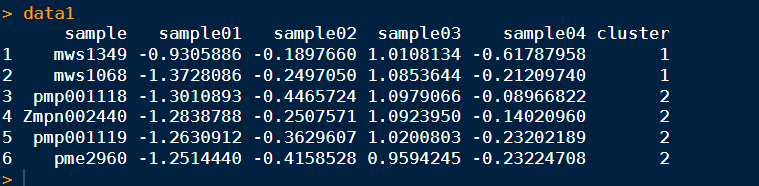
cluster 是我们已经做好分类的列。
data2 <- pivot_longer(data1, cols = -c(sample, cluster), names_to = "group", values_to = "value")data2 <- data2[, c("sample", "value", "group", "cluster")]
data2df2 %
group_by(cluster, group) %>%
summarise(value = mean(value))
df2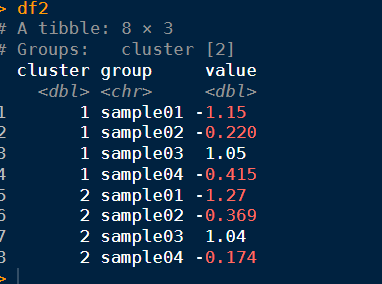
ggplot(data2, aes(x = group, y = value)+)
gm_line(aes(group = sample), color = "grey90", size = 0.5)+
##'@X轴因子固定,结合自己的数据进行修改
scale_x_discrete(limits = c("sample1", "sample2", "sample3", "sample4", "sample5")) +
geom_line(data = df2, aes(x = group, y = value, group = 1), color = "red", size = 1)+
facet_wrap(~ factor(cluster), nrow = 2)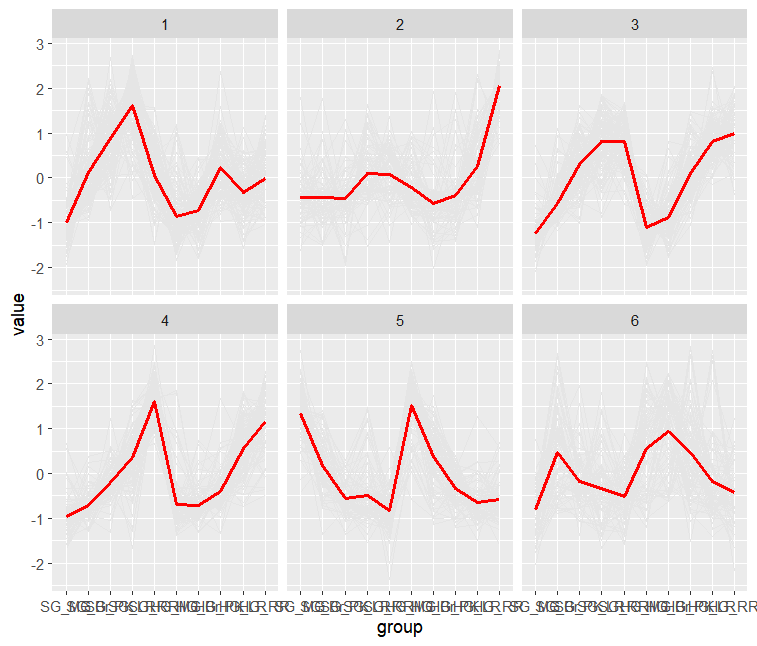
ggplot(data2, aes(x = group, y = value)+)
gm_line(aes(group = sample), color = "grey90")+
##'@X轴因子固定,结合自己的数据进行修改
scale_x_discrete(limits = c("sample1", "sample2", "sample3", "sample4", "sample5"))+
stat_summary(aes(group = 1), fun.y = "mean", geom = "line", size = 1, color = "red")+
theme_classic(base_size = 14)+
theme(axis.ticks.length = unit(0.1,'cm'),
axis.text.x = element_text(angle = 45,
hjust = 1,color = 'black'),
strip.background = element_blank())+
facet_wrap(~factor(cluster), nrow = 2)+
ylab('Normalized expression') + xlab(NULL)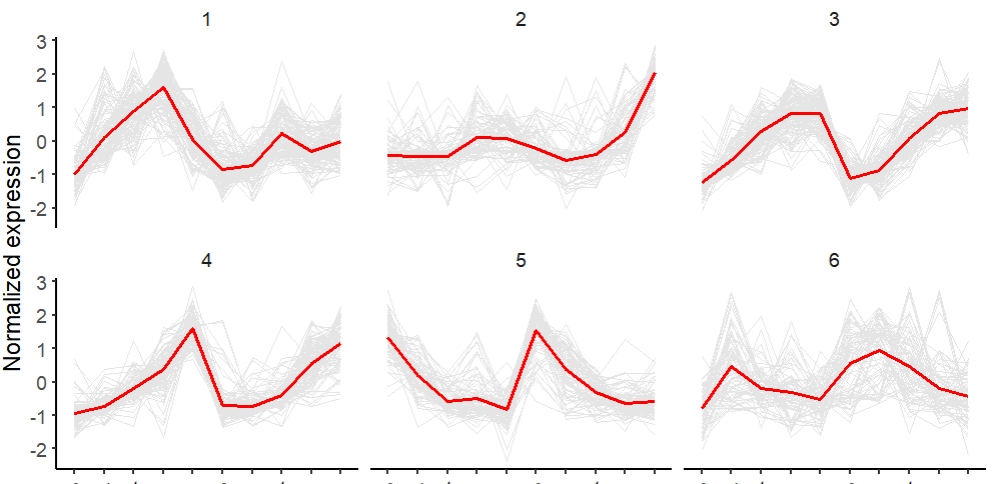
图形其余美化,结合自己的需求进行调整即可。
聚类分析在基因表达数据中可以用于鉴定共表达基因,这对于理解基因的功能和调控网络至关重要。
通过聚类分析,我们可以将表达模式相似的基因聚为一类,进而分析这些基因的生物学功能。
在蛋白质组学中,聚类分析同样重要,可以帮助我们识别参与相同生物学过程的蛋白质。
通过分析蛋白质表达模式的聚类,我们可以识别在特定生物学过程中起关键作用的蛋白质。
答:K-means聚类是一种将数据点分成K个簇的算法,使得簇内的点尽可能相似,簇间的点尽可能不同。
答:选择合适的K值可以通过多种方法,如肘部法则、轮廓系数等,这些方法可以帮助我们评估不同K值下的聚类效果。
答:K-means聚类的优点包括简单、高效,适用于大规模数据集;缺点包括对初始质心敏感,可能需要多次运行以获得稳定结果,且对噪声和离群点敏感。
答:处理离群点可以采用多种策略,如在聚类前预处理数据以识别和移除离群点,或者使用对离群点不敏感的聚类算法,如K-medoids。
答:聚类分析在生物信息学中有广泛的应用,包括基因表达数据的分析、蛋白质组学数据的分析、代谢组学数据的分析等,可以帮助我们识别共表达的基因、关键蛋白质和重要代谢物。





